


Cerebellar liponeurocytoma is a rare, low-grade neoplasm (WHO grade 2) characterized by neuronal or neurocytic differentiation and variable glial differentiation with lipoma-like alterations. First described in 1978 and recognized in the WHO classification in 2000, it presents significant challenges in radiological and pathological differentiation. This differentiation is crucial to avoid unnecessary or deleterious treatments.
Observations
The patient presented with symptoms including headache, dizziness, hiccups, nausea, vomiting, tinnitus, and left-sided ataxia. Imaging revealed a heterogeneous lesion in the left cerebellopontine angle. Resection was performed via a left suboccipital retrosigmoid approach, resulting in significant improvement in axial and appendicular ataxia, although left-sided anacusis persisted. Histopathological analysis confirmed cerebellar liponeurocytoma showing hypercellularity, with neurocytic cells containing lipid accumulations. Immunohistochemical analysis revealed diffuse expression of synaptophysin and focal expression of glial fibrillary acidic protein, with a Ki-67 proliferation index of 7%.
Lessons
Cerebellar liponeurocytoma is a rare tumor requiring accurate histopathological differentiation to determine the appropriate treatment. Resection remains the mainstay of treatment, with postoperative radiotherapy potentially reducing recurrence. Multidisciplinary follow-up is crucial for managing residual symptoms and monitoring for recurrence. This case aligns with existing literature and underscores the importance of comprehensive diagnostic and therapeutic approaches to improve patient outcomes.
Keywords
cerebellar liponeurocytoma; CNS tumor; rare tumor; case report
Introduction
Cerebellar liponeurocytoma is a rare, low-grade neoplasm (WHO grade 2) characterized by neuronal or neurocytic differentiation, as well as variable glial differentiation with lipoma-like alterations. This tumor of neuroectodermal origin was first described by Bechtel et al. in 1978 as a mixed mesenchymal and neuroectodermal tumor. It was only in 2000 that it was recognized in the WHO classification under the name “cerebellar liponeurocytoma,” emphasizing its neurocytic differentiation, and was classified within the group of grade I neuronal tumors. In 2016, WHO reclassified it as a grade II neurocytic/neural tumor, and finally, in the 2021 classification, it was categorized within the group of glioneuronal tumors. Other terminologies previously used to refer to liponeurocytoma include “lipomatous or lipidized medulloblastoma,” “medullocytoma,” and “lipomatous glioneurocytoma,” which are not recommended by WHO.
There are a little more than 70 reported cases in the literature, and the tumor represents a challenge in its radiological and pathological differentiation. This differentiation is crucial to avoid unnecessary or even deleterious adjunctive treatments and therapies for the patient.
Illustrative Case
A 60-year-old male patient presented with a 15-day history of headache, dizziness, hiccups, nausea, and vomiting, accompanied by tinnitus in the left ear. Additionally, he reported removal of a bone tumor from the right lower limb 10 years prior, with no available histopathological details.
On seeking emergency care, the patient underwent physical examination, which revealed multidirectional nystagmus, left-sided anacusis, left-sided axial and appendicular ataxia with a significant ataxic gait, and right-sided tactile and pain hyperesthesia. Additionally, the patient exhibited a left-sided facial nerve paralysis, classified as House-Brackmann grade II. The patient denied any other previous symptoms. An initial cranial CT scan demonstrated a solid-cystic lesion in the left cerebellopontine angle, which was further evaluated using MRI. The preoperative MRI revealed an expansive extra-axial lesion in the left cerebellopontine cistern, extending into the internal acoustic canal, with lobulated contours. The lesion displayed intermediate signal intensity on T1-weighted images and heterogeneous hyperintensity on T2-weighted images, with no diffusion restriction. Intense and heterogeneous enhancement was observed following administration of a paramagnetic contrast agent, with the lesion measuring approximately 5.1 × 4.1 × 3.2 cm. Peripheral cystic areas exhibited hypointensity on T1-weighted images, hyperintensity on T2-weighted images, and no enhancement after contrast. The lesion caused compression and contralateral displacement of the pons and the left middle cerebellar peduncle, reducing the fourth ventricle and leading to dilation of the supratentorial ventricular system.
The patient underwent resection of the lesion via a left suboccipital retrosigmoid approach. Neurophysiological intraoperative monitoring was used. The tumor was identified as a predominantly extra-axial lesion with a good cleavage plane; however, it demonstrated invasion into the internal acoustic canal. As a result, the canal was opened during surgery to ensure adequate tumor resection. The medial portion of the tumor lacked a clear plane of cleavage with the brainstem, complicating complete resection. Tumor invasion into the jugular foramen necessitated manipulation of this region during surgery. The cranial nerves were found to be displaced anteriorly and inferiorly by the tumor. Microsurgical techniques were used to resect the tumor, which presented as dark reddish, mostly soft, and well vascularized. Intraoperatively, significant decreases in the potentials of left cranial nerves VII and XI were observed during monitoring, but no changes in cranial nerve X monitoring were detected. Postoperatively, the patient presented with left-sided facial nerve paralysis, progressing from House-Brackmann grade II preoperatively to grade IV immediately postoperatively. Additionally, the patient exhibited alterations in palatal function, attributed to left-sided cranial nerve X dysfunction, likely due to manipulation in the jugular foramen region.
Within 48 hours of surgery, the patient developed signs of hydrocephalus, requiring placement of a ventriculoperitoneal shunt. The patient experienced significant improvement in axial and appendicular ataxia, while left-sided anacusis persisted. Over the following weeks, facial nerve function improved to House-Brackmann grade III. Follow-up cranial MRI at 6 months postoperatively revealed no signs of tumor recurrence, only leptomeningeal thickening, possibly due to cerebrospinal fluid hypotension. Functionally, the patient has shown improvement in gait and facial palsy but continues to experience left-sided anacusis. The patient also has vocal fold palsy and is being followed by the speech therapy team, without signs of dysphagia on videoendoscopy. After tumor resection, the patient underwent 28 sessions of radiotherapy, totaling 5040 cGy. He continues to be monitored by multidisciplinary rehabilitation and oncology teams.
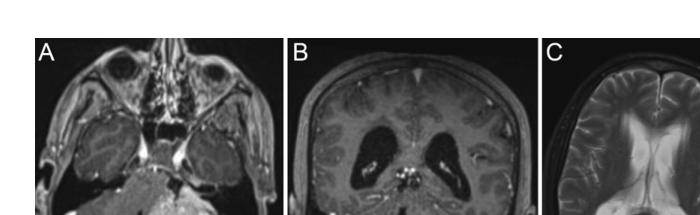
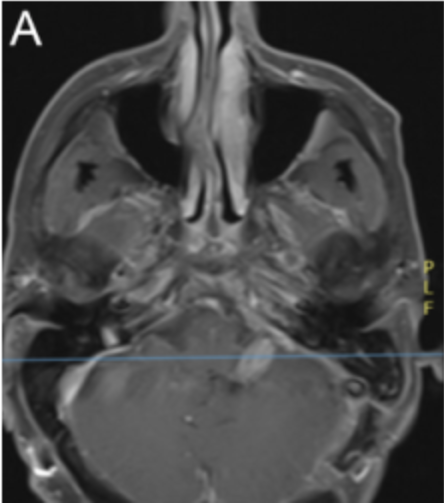
Figure 1. Preoperative MRI showing a contrast-enhancing lesion in the left cerebellopontine angle with a solid area obliterating the fourth ventricle, coronal tumor view, and ependymal transudation caused by hydrocephalus.
Informed Consent
The necessary informed consent was obtained in this study.
Discussion
Cerebellar liponeurocytoma predominantly occurs in the cerebellar hemispheres but can also involve the cerebellar vermis and the cerebellopontine angle, as demonstrated in this clinical case. Some case series have reported liponeurocytoma-like tumors in supratentorial regions, which can be confused with central or extraventricular neurocytomas with lipomatous changes. There are also reports of multiple spinal cord liponeurocytomas and familial associations.
The average age of affected patients is 50 years, with a peak incidence between the 3rd and 6th decades of life, showing no sex predilection. There might be an association with autosomal dominant familial inheritance. Currently, more than 70 cases have been described in the literature, underscoring the rarity of this tumor.
The most common clinical presentations of the tumor are symptoms related to the posterior fossa: gait disturbances, imbalance and motor coordination issues, nausea, vomiting, and hearing loss. Additionally, compression of the fourth ventricle can result in headaches due to hydrocephalus with signs of intracranial hypertension. This patient exhibited all these symptoms.
Differential diagnoses include oligodendroglioma, ependymoma, and medulloblastoma. Differentiation is based on histopathological findings: oligodendroglioma is positive for the Olig2 marker, whereas ependymoma and medulloblastoma are negative for the EMA marker. Furthermore, 20% of the lesions show mutations in the p53 tumor protein, APC gene, and KDR gene.
In comparison with the cases in the existing literature, this case aligns with the typical presentation of cerebellar liponeurocytoma, including its localization in the cerebellopontine angle and its clinical manifestations. This supports the finding that cerebellar liponeurocytoma, although rare, can present with a constellation of symptoms and diagnostic challenges consistent with previously described case reports.
Total tumor resection is the most commonly used treatment, and due to the rarity of the disease, there is no well-defined protocol. It is indicated for all patients with clinical and radiological suspicion of cerebellar liponeurocytoma. The most commonly used surgical approaches are retrosigmoid and median suboccipital craniotomies. The choice of craniotomy depends on the tumor’s location.
Intraoperative findings typically include a well-circumscribed, red-yellowish-gray, soft-consistency, well-vascularized, and infiltrative lesion. The most common complication is hemorrhage due to the high vascularization of the tumor. Adjuvant radiation therapy is recommended in approximately 70% of cases, primarily when there is a higher risk of recurrence associated with subtotal resection, where tumor tissue remains. In contrast, about 30% of cases, generally involving gross-total resection, do not receive adjuvant radiation therapy, as complete tumor removal significantly reduces recurrence risk. Therefore, the decision to use adjuvant radiation depends on the extent of resection and individual patient risk factors.
Cerebellar liponeurocytomas typically appear as heterogeneous intra-axial masses or, less commonly, intraventricular masses. The most common location is the cerebellar hemispheres. On MRI, the primary findings include a well-defined, heterogeneous intra-axial lesion with significant edema and neovascularization, most commonly located in the cerebellar hemispheres or, less frequently, intraventricularly.
Histopathologically, a cerebellar liponeurocytoma is composed of a uniform population of small neurocytic cells arranged in lobules or diffuse architecture, exhibiting rounded or oval nuclei, clear cytoplasm, and poorly defined cell borders. The hallmark histological feature is the presence of neuroepithelial cells with intracellular lipid accumulation resembling adipocytes. Typically, there is no anaplasia, and primary lesions present with a low Ki-67 index (< 4%). Immunohistochemically, the tumor shows expression of neuronal markers such as synaptophysin, NeuN, and MAP2. Focal expression of glial fibrillary acidic protein indicates astrocytic differentiation.
Genetic profiling has provided evidence that cerebellar liponeurocytomas are a distinct clinical entity more closely related to neurocytomas than to medulloblastomas. Overexpression of fatty acid-binding protein 4 can help distinguish these tumors from medulloblastomas. Genome-wide analyses have identified focal and recurrent loss of chromosome 16 and 2p. This genetic expression profile is aligned more with neurocytomas than with medulloblastomas. In some cases, cerebellar liponeurocytomas can exhibit TP53 missense mutations at a much higher frequency than medulloblastomas, a mutation not found in neurocytomas.
The histopathological analysis in this patient revealed hypercellularity composed of small neurocytic cells arranged in diffuse sheets, which exhibit round or oval nuclei and poorly defined clear cytoplasm. Notably, there was an accumulation of lipids within the cell cytoplasm, resembling adipocytes. There was no evidence of anaplasia, necrosis, or microvascular proliferation. Immunohistochemical analysis showed a diffuse expression of synaptophysin, focal expression of glial fibrillary acidic protein, and a Ki-67 proliferation index of 7%.
The prognosis for cerebellar liponeurocytoma is uncertain due to the high recurrence rate. Some studies have shown that patients who underwent total tumor resection and received postoperative radiotherapy had a reduced chance of recurrence.
Follow-up cranial MRI in January 2024 showed no signs of tumor recurrence, only leptomeningeal thickening, possibly due to cerebrospinal fluid hypotension. The patient continues to rely on a ventriculoperitoneal shunt. Functionally, the patient showed improvement in gait and facial palsy. The patient also had vocal fold palsy and was being followed by the speech therapy team, without signs of dysphagia on videoendoscopy.
After tumor resection, the patient underwent 28 sessions of radiotherapy. He continues to be monitored by multidisciplinary rehabilitation and oncology teams.
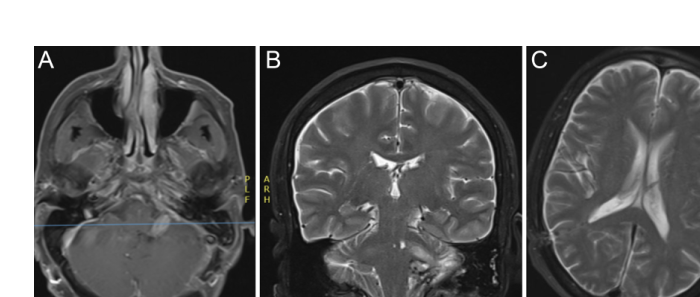
Figure 2. Postoperative MRI at 6 months after resection, demonstrating resolution of hydrocephalus and minimal contrast uptake near the left sigmoid sinus region.
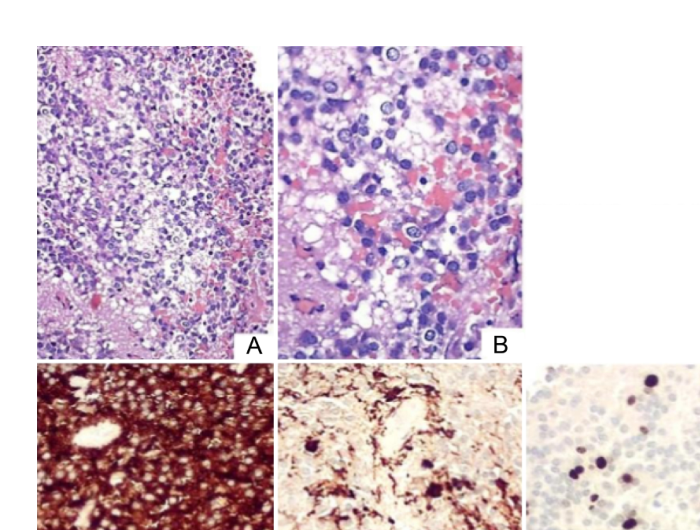
Figure 3. Histopathological findings: small neurocytic cells with focal accumulation resembling adipocytes, diffuse expression of synaptophysin, focal expression of GFAP, and Ki-67 proliferation index of 7%.
Final Lessons
Cerebellar liponeurocytoma is a rare, low-grade entity that typically affects patients around 50 years old, with no sex predilection and a possible familial association. Currently, there are more than 70 cases described in the literature. Accurate histopathological differentiation is crucial for determining the appropriate adjuvant treatment to improve the prognosis.
Halisson R. de Andrade, MD; Fernanda Fenner, MD; Valentina Pochio Vasques; Mariana Matos Vasconcelos, MD; Marcio S. Rassi, MD, PhD; Jean G. de Oliveira, MD, PhD; Carmen Lúcia Penteado Lancellotti, MD, PhD
Division of Neurosurgery, Department of Surgery, Santa Casa de São Paulo School of Medical Sciences, São Paulo, Brazil; Santa Casa de São Paulo School of Medical Sciences, São Paulo, Brazil; Pathology Department, Santa Casa de São Paulo School of Medical Sciences, São Paulo, Brazil.
Journal of Neurosurgery: Case Lessons
Published March 17, 2025
DOI: 10.3171/CASE24521
License: CC BY-NC-ND 4.0
References
1. WHO Classification of Tumours: Central Nervous System Tumours. Vol 6. 5th ed. IARC Publications; 2021.
2. Oudrhiri MY, Raouzi N, El Kacemi I, et al. Understanding cerebellar liponeurocytomas: case report and literature review. Case Rep Neurol Med. 2014;2014:186826.
3. Bechtel JT, Patton JM, Takei Y. Mixed mesenchymal and neuroectodermal tumor of the cerebellum. Acta Neuropathol. 1978;41(3):261-263.
4. Louis DN, Perry A, Wesseling P, et al. The 2021 WHO classification of tumors of the central nervous system: a summary. Neuro Oncol. 2021;23(8):1231-1251.
5. Louis DN, Perry A, Reifenberger G, et al. The 2016 World Health Organization classification of tumors of the central nervous system: a summary. Acta Neuropathol. 2016;131(6):803-820.
6. Gembruch O, Junker A, Mönninghoff C, et al. Liponeurocytoma: systematic review of a rare entity. World Neurosurg. 2018;120:214-233.
7. Wolf A, Alghefari H, Krivosheya D, et al. Cerebellar liponeurocytoma: a rare intracranial tumor with possible familial predisposition. Case report. J Neurosurg. 2016;125(1):57-61.
8. Zuo P, Sun T, Gu G, et al. Surgical management and clinical outcomes of cerebellar liponeurocytomas – a report of seven cases and a pooled analysis of individual patient data. Neurosurg Rev. 2022;45(2):1747-1757.
9. Ali R, Durrani S, Nathani KR, Jarrah R, Bydon M. Cerebellar liponeurocytoma: publication trends, scientometrics analysis, and critical review. World Neurosurg. 2023;171:e137-e146.
10. Wang S, Xu X, Wang C. Radiological and clinical findings of multiple cerebellar liponeurocytoma: a case report. Front Surg. 2021;8:686892.
11. Wang KE, Ni M, Wang L, et al. Cerebellar liponeurocytoma: a case report and review of the literature. Oncol Lett. 2016;11(2):1061-1064.
12. Abuzneid YS, Alzeerelhouseini HIA, Shkokani S, Aqel W, Aldarawish A. Cerebellar liponeurocytoma, a rare tumor: case report and review of the literature. Int J Surg Case Rep. 2021;82:105937.
13. Radke J, Gehlhaar C, Lenze D, et al. The evolution of the anaplastic cerebellar liponeurocytoma: case report and review of the literature. Clin Neuropathol. 2015;34(1):19-25.
14. Giangaspero F, Cenacchi G, Roncaroli F, et al. Medullocytoma (Lipidized medulloblastoma). A cerebellar neoplasm of adults with favorable prognosis. Am J Surg Pathol. 1996;20(6):656-664.
15. Soylemezoglu F, Soffer D, Onol B, Schwechheimer K, Kleihues P. Lipomatous medulloblastoma in adults. A distinct clinicopathological entity. Am J Surg Pathol. 1996;20(4):413-418.
16. Horstmann S, Perry A, Reifenberger G, et al. Genetic and expression profiles of cerebellar liponeurocytomas. Brain Pathol. 2004;14(3):281-289.
17. Anghileri E, Eoli M, Paterra R, et al. FABP4 is a candidate marker of cerebellar liponeurocytomas. J Neurooncol. 2012;108(3):513-519.
18. Capper D, Stichel D, Sahm F, et al. Practical implementation of DNA methylation and copy-number-based CNS tumor diagnostics: the Heidelberg experience. Acta Neuropathol. 2018;136(2):181-210.
Disclosures
The authors report no conflict of interest concerning the materials or methods used in this study or the findings specified in this paper.
Author Contributions
Conception and design: de Andrade, Fenner, de Oliveira, Lancellotti.
Acquisition of data: de Andrade, Fenner, Rassi, Lancellotti.
Analysis and interpretation of data: de Andrade, Fenner, Vasconcelos, Lancellotti.
Drafting the article: de Andrade, Fenner, Vasques, Vasconcelos.
Critically revising the article: Fenner, Rassi, de Oliveira, Lancellotti.
Reviewed submitted version of manuscript: de Andrade, Fenner, Rassi, de Oliveira, Lancellotti.
Approved the final version of the manuscript on behalf of all authors: de Andrade.
Administrative/technical/material support: de Andrade, Rassi, Lancellotti.
Study supervision: de Andrade, Rassi, de Oliveira, Lancellotti.
Correspondence
Halisson R. de Andrade: Santa Casa de São Paulo School of Medical Sciences, São Paulo, Brazil. deandrade.hr@gmail.com.